Latest News
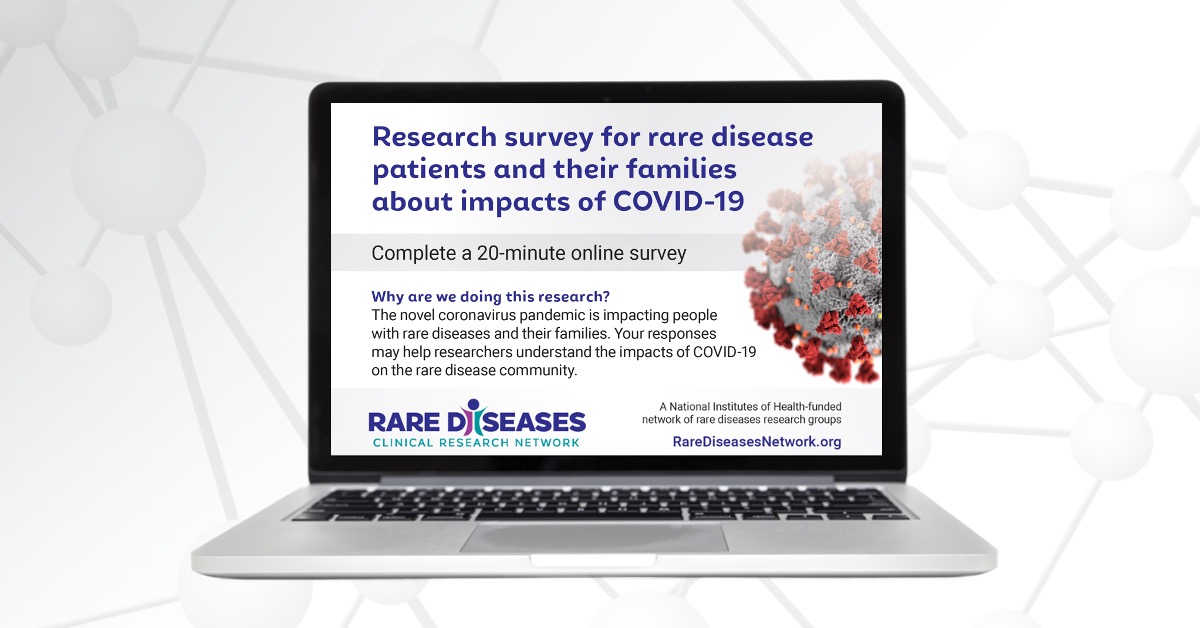
National Survey Reveals Impact of COVID-19 on People Living with Rare Diseases and Their Families
The devastating impact of COVID-19 on the general population is well-documented—but less is known about the millions of people living with rare diseases.